
Nowe leki w praktyce: co, dla kogo i kiedy w Polsce?
Streszczenie
W ostatnich latach pojawiła się fala przełomowych terapii w różnych dziedzinach medycyny – od chorób neurodegeneracyjnych, przez onkologię, po choroby rzadkie. Wiele z tych leków to pierwsze w swojej klasie lub pierwsze skuteczne terapie dla dotychczas nieuleczalnych schorzeń. Pomimo dopuszczenia tych terapii do obrotu w Polsce, często wciąż pozostają one niedostępne dla pacjentów ze względu na brak objęcia refundacją przez Narodowy Fundusz Zdrowia. Odpowiedzią na brak refundacji mogą być jednak mechanizmy nadzwyczajne: import docelowy, Ratunkowy Dostęp do Technologii Lekowych (RDTL) oraz programy wczesnego dostępu.
Słowa kluczowe
innowacyjne terapie, programy wczesnego dostępu, Ratunkowy Dostęp do Technologii Lekowych (RDTL), import docelowy
Wprowadzenie
Postęp w medycynie wchodzi w etap, w którym innowacje przestają być incydentalne, a stają się zjawiskiem systemowym. Zbieg kilku trendów – od rutynowego sekwencjonowania genomu, przez inżynierię białek i komórek, po edycję genów i lepsze biomarkery – radykalnie skrócił drogę od odkrycia do terapii. Równolegle regulatorzy (m.in. EMA i FDA) wdrożyli przyspieszone ścieżki oceny i narzędzia doradcze, co ułatwia wejście leków o wysokiej wartości klinicznej. Wspólnym mianownikiem tych terapii jest precyzyjne celowanie w mechanizm choroby, często potwierdzone kryteriami molekularnymi i mierzalnymi punktami końcowymi. Co ważne, ta zmiana nie pozostaje teorią – już przekłada się na konkretne interwencje dostępne (lub wchodzące) do praktyki klinicznej.
W tym kontekście w ostatnich latach pojawiła się fala przełomowych terapii w różnych dziedzinach medycyny – od chorób neurodegeneracyjnych, przez onkologię, po choroby rzadkie (Tabela 1). Wiele z tych leków to pierwsze w swojej klasie lub pierwsze skuteczne terapie dla dotychczas nieuleczalnych schorzeń. Przykładowo, donanemab to przeciwciało monoklonalne usuwające patogenny amyloid β, stosowane w celu spowolnienia przebiegu choroby Alzheimera u pacjentów we wczesnej fazie, lifileucel to pierwsza terapia oparta o limfocyty infiltrujące guz (TIL) zatwierdzona w onkologii, exagamglogene autotemcel jest pierwszą klinicznie zatwierdzoną terapią genową wykorzystującą edycję CRISPR/Cas9, zaś atidarsagene autotemcel to pierwsza terapia genowa dla dzieci z leukodystrofią metachromatyczną (MLD) zapobiegająca neurodegeneracji. W onkologii pojawiły się terapie celujące w konkretne nieprawidłowości molekularne: zenocutuzumab – pierwszy lek ukierunkowany na nowotwory z fuzją genu NRG1; inavolisib – inhibitor PI3Kα stosowany przy mutacjach PIK3CA; oraz tarlatamab – nowa immunoterapia (przeciw DLL3) dla opornego drobnokomórkowego raka płuca. Z kolei w endokrynologii crinecerfont to pierwszy od dekad lek dla wrodzonego przerostu nadnerczy (antagonista receptora CRF1, pozwalający zmniejszać dawki steroidów). Lista obejmuje też innowacyjne terapie RNA (olezarsen – nowa opcja w rodzinnej chylomikronemii, ukierunkowana na metabolizm lipidów), nowe leki w chorobach autoimmunologicznych (nemolizumab – przeciwciało blokujące receptor IL-31 stosowane w leczeniu świerzbiączki) oraz długo oczekiwany resmetirom – pierwszy lek na NASH z włóknieniem.
W artykule wyjaśniamy, w jakim stopniu te leki są już dostępne w Europie i w Polsce oraz jakie mechanizmy mogą zapewnić pacjentom dostęp do terapii ratujących życie.
Tabela 1. Status i dostępność wybranych terapii innowacyjnych.
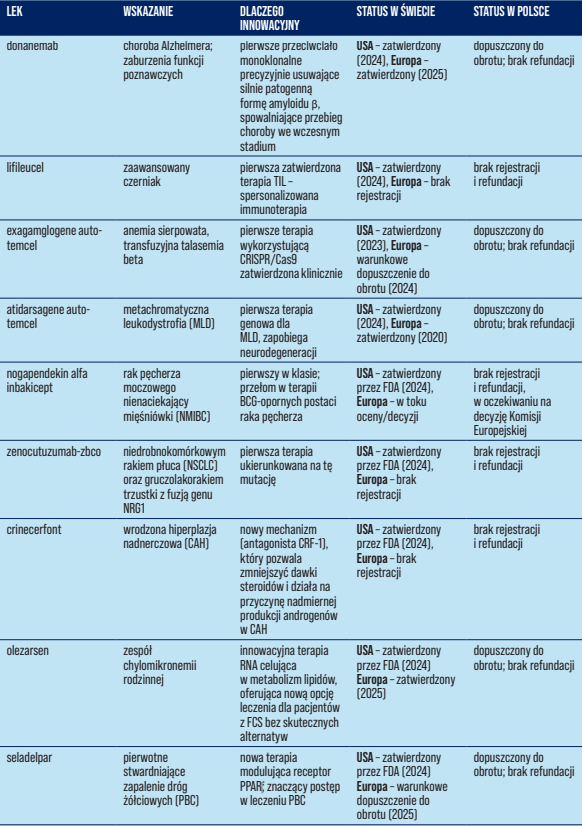
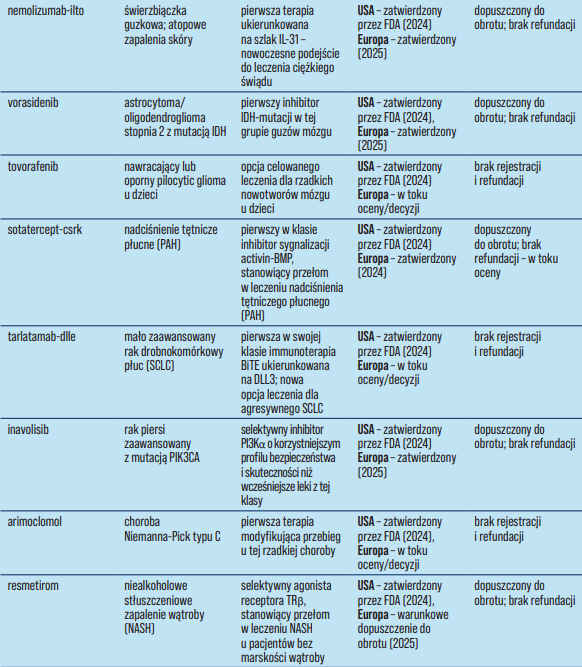
Status rejestracji w USA i Europie
Większość omawianych terapii uzyskała zatwierdzenie Amerykańskiej Agencji FDA w 2024 r. W Stanach Zjednoczonych wiele z nich otrzymało tryb przyspieszonej procedury (accelerated approval) lub status przełomowej terapii (breakthrough therapy) z uwagi na wysoki poziom niezaspokojonych potrzeb klinicznych oraz przełomowy charakter działania.
Europa również stopniowo dopuszcza te terapie, zwykle z kilkumiesięcznym lub nawet rocznym opóźnieniem względem decyzji FDA. Do jesieni 2025 r. w Unii Europejskiej zatwierdzono już m.in.: exagamglogene autotemcel, atidarsagene autotemcel, seladelpar, nemolizumab, inavolisib, sotatercept, olezarsen, vorasidenib, resmetirom oraz donanemab.
Część pozostałych terapii wciąż znajduje się na etapie oceny w EMA (np. arimoclomol, tovorafenib, tarlatamab, nogapendekin alfa inbakicept), a niektóre nie mają jeszcze złożonego wniosku rejestracyjnego. Warto podkreślić, że uzyskanie unijnego pozwolenia to dopiero pierwszy krok – realny dostęp wymaga jeszcze krajowych decyzji refundacyjnych i negocjacji cenowych.
Dostępność w Polsce i perspektywy refundacji
W Polsce, zgodnie z prawem farmaceutycznym Unii Europejskiej, leki zatwierdzone w procedurze centralnej przez Komisję Europejską są automatycznie dopuszczone do obrotu również na terytorium Rzeczypospolitej Polskiej. Oznacza to, że jeśli dana terapia uzyskała pozwolenie na dopuszczenie do obrotu wydane przez Europejską Agencję Leków oraz Komisję Europejską, może być legalnie sprzedawana i stosowana także w naszym kraju. Sama rejestracja nie oznacza jednak automatycznej dostępności dla pacjentów. Kluczowym warunkiem realnego dostępu do terapii jest objęcie jej finansowaniem ze środków publicznych, czyli refundacją przez Narodowy Fundusz Zdrowia.
Bez refundacji pacjent musiałby pokryć pełny koszt terapii samodzielnie, co w przypadku nowoczesnych, innowacyjnych leków jest praktycznie niewykonalne – ceny często sięgają setek tysięcy złotych, a w przypadku jednorazowych terapii genowych nawet kilku milionów złotych. Obecnie żaden z nowo zarejestrowanych leków innowacyjnych nie został jeszcze objęty refundacją w Polsce, mimo że część z nich uzyskała już rejestrację w Unii Europejskiej. W wielu przypadkach producenci nie złożyli nawet wniosków refundacyjnych. Wynika to z faktu, że Polska rzadko bywa traktowana przez firmy farmaceutyczne jako rynek priorytetowy – pierwszeństwo mają zazwyczaj takie kraje jak Niemcy, Francja czy Wielka Brytania. Producenci stosują strategię tzw. sekwencyjnego wprowadzania leków, aby ograniczać ryzyko niekorzystnego oddziaływania zewnętrznej referencji cenowej, co naturalnie opóźnia moment złożenia wniosku w Polsce i tym samym przesuwa perspektywę refundacji.
Dane Europejskiej Federacji Przemysłu i Stowarzyszeń Farmaceutycznych wskazują, że średni czas uzyskania dostępu do innowacyjnego leku w Europie wynosi około 531 dni. W Polsce okres ten jest jednym z najdłuższych w całej Unii Europejskiej – średnio ponad 800 dni od momentu rejestracji do realnego dostępu dla pacjenta [1]. Od tej reguły zdarzają się jednak wyjątki. W przypadku terapii sotaterceptem, stosowanej w leczeniu tętniczego nadciśnienia płucnego, postępowanie refundacyjne jest w toku – Rada Przejrzystości wydała pozytywne stanowisko dla czterech prezentacji leku w programie lekowym, lecz wciąż brak ostatecznej decyzji. Tworzy to realną perspektywę objęcia terapii programem w najbliższym czasie, co miałoby duże znaczenie dla pacjentów z tą ciężką i rzadką chorobą.
Podsumowując, obecnie żaden z omawianych leków innowacyjnych nie jest w Polsce dostępny w rutynowej praktyce klinicznej w ramach refundacji. Część z nich jest co prawda zarejestrowana i dopuszczona do obrotu, lecz brak finansowania ze środków publicznych sprawia, że w praktyce są poza zasięgiem zdecydowanej większości chorych. Inne leki wciąż czekają na rejestrację unijną i do tego czasu nie mogą być standardowo stosowane. W rezultacie polski pacjent, u którego zawiodły standardowe metody leczenia, staje przed poważnym wyzwaniem – jak uzyskać dostęp do nowoczesnej terapii, zanim stanie się ona szeroko dostępna w ramach refundacji. Odpowiedź na to pytanie stanowią mechanizmy wcześniejszego dostępu, które omówione są w dalszej części artykułu.
Możliwości wcześniejszego dostępu w Polsce
Choć omawiane leki nie są obecnie refundowane, polski system przewiduje mechanizmy umożliwiające pacjentom dostęp do terapii jeszcze przed formalnym finansowaniem lub nawet przed rejestracją. Należą do nich: program wczesnego dostępu (compassionate use), Ratunkowy Dostęp do Technologii Lekowych (RDTL) oraz import docelowy.
Compassionate use (program wczesnego dostępu) to zorganizowane, wielopacjenckie udostępnianie leku przez producenta przed uzyskaniem pozwolenia na dopuszczenie do obrotu i przed refundacją. Stosuje się go u chorych z ciężkimi, zagrażającymi życiu lub przewlekle wyniszczającymi schorzeniami, gdy wyczerpano dostępne terapie i pacjent nie kwalifikuje się do badania klinicznego. Po uzgodnieniu z organami regulacyjnymi producent uruchamia program z jasno opisanymi kryteriami kwalifikacji oraz listą ośrodków; lek jest zwykle dostarczany nieodpłatnie, natomiast ośrodek pokrywa koszty standardowej opieki i monitoringu bezpieczeństwa. Mechanizm ten funkcjonuje w „oknie” pomiędzy zakończeniem kluczowych badań a formalnymi decyzjami rejestracyjnymi i płatniczymi.
Podstawą prawną na poziomie UE jest art. 83 rozporządzenia (WE) 726/2004 (w polskiej wersji określany jako „indywidualne stosowanie”), który przewiduje opinie CHMP/EMA dotyczące warunków stosowania i dystrybucji, przy czym wdrożenie pozostaje w gestii państw członkowskich i producenta. EMA publikuje wybrane opinie, ale nie prowadzi pełnego centralnego wykazu aktywnych programów – w praktyce najpewniejszym źródłem aktualnych informacji jest sam producent (sekcje „Medical Information”, „Early/Compassionate Access/EAP”). Należy pamiętać, że uruchomienie programu zależy od woli firmy, zasięg bywa geograficznie ograniczony, a kryteria medyczne są ściśle zdefiniowane.
W przypadku braku programu wczesnego dostępu alternatywą dla pojedynczych pacjentów jest import docelowy. To indywidualna procedura umożliwiająca sprowadzenie z zagranicy produktu leczniczego niedopuszczonego do obrotu w Polsce i bez dostępnego odpowiednika na rynku krajowym, jeżeli lekarz uzna, że jest on niezbędny dla konkretnego pacjenta i brak jest skutecznych alternatyw terapeutycznych. Wniosek przygotowuje lekarz (z uzasadnieniem medycznym) i przekazuje go – wraz z opinią właściwego konsultanta (krajowego lub wojewódzkiego) – do Ministra Zdrowia. Po uzyskaniu zgody lek może zostać sprowadzony przez uprawnioną hurtownię farmaceutyczną i wydany pacjentowi zgodnie ze wskazaniami określonymi we wniosku. Co do zasady, import docelowy nie zapewnia automatycznej refundacji: koszty ponosi pacjent lub szpital, chyba że Minister Zdrowia wyda odrębną zgodę na finansowanie. Procedura bywa czasochłonna i wymaga ścisłej współpracy lekarza, pacjenta oraz podmiotu sprowadzającego. Należy podkreślić, że realizacja importu zależy od zgody producenta / podmiotu odpowiedzialnego, który może odmówić jej udzielenia także bez podania przyczyny.
Jeżeli lek został już dopuszczony do obrotu w Unii Europejskiej lub w Polsce, ale nie uzyskał jeszcze refundacji, istnieje alternatywny mechanizm wsparcia dla pacjentów – Ratunkowy Dostęp do Technologii Lekowych (RDTL). Umożliwia on szpitalowi uzyskanie publicznego finansowania terapii dla konkretnego pacjenta znajdującego się w stanie zagrożenia życia lub zdrowia, gdy brak jest skutecznych metod leczenia. Procedura ma charakter scentralizowany. Lekarz składa wniosek wraz z uzasadnieniem i opinią konsultanta krajowego lub wojewódzkiego w danej dziedzinie, a ostateczną decyzję podejmuje Minister Zdrowia. Po jej wydaniu Narodowy Fundusz Zdrowia może pokryć koszty terapii na okres 3 miesięcy, z możliwością przedłużenia finansowania w przypadku potwierdzonej skuteczności leczenia. W praktyce RDTL pełni rolę „pomostu” – pozwala pacjentom uzyskać dostęp do terapii już zarejestrowanych w UE, ale jeszcze nierefundowanych w Polsce. Dzięki temu możliwe jest wcześniejsze wprowadzenie innowacyjnych leków, zanim zostaną objęte programami lekowymi lub refundacją standardową. Istotnym ograniczeniem mechanizmu jest jednak budżet. Wysokokosztowe terapie, takie jak jednorazowe terapie genowe warte kilka milionów złotych, mogą być trudne do sfinansowania w ramach RDTL, ponieważ ich koszt mógłby znacząco obciążyć dostępne środki. Niemniej jednak procedura ta daje realną możliwość leczenia pacjentów w sytuacjach krytycznych. W przeszłości RDTL był często wykorzystywany do finansowania nowoczesnych terapii onkologicznych u pacjentów, u których wyczerpano inne refundowane metody leczenia.
Podsumowując, Polska znajduje się na końcu ścieżki dostępu do nowych terapii – po zatwierdzeniu w USA i UE pacjenci w kraju muszą czekać na refundację. Obecnie żadna z omawianych terapii nie jest refundowana w Polsce, co tworzy tzw. lukę innowacyjną. Inne kraje UE szybciej udostępniają te leki, często w ramach specjalnych programów lub funduszy. W Polsce istnieją jednak mechanizmy nadzwyczajne: import docelowy, Ratunkowy Dostęp do Technologii Lekowych (RDTL) oraz compassionate use. Choć ograniczone, stworzyły pacjentom możliwość skorzystania z nowoczesnych terapii.
Bibliografia
- European Federation of Pharmaceutical Industries and Associations (EFPIA).Patients W.A.I.T. Indicator Survey 2024. EFPIA; 2024.
Oleksandr Shtyka
Koordynator ds. Współpracy z Zagranicą
Dział Współpracy z Zagranicą, Urtica Sp. z o.o.